Pseudopotentials
In this example, we'll look at how to use various pseudopotential (PSP) formats in DFTK and discuss briefly the utility and importance of pseudopotentials.
Currently, DFTK supports norm-conserving (NC) PSPs in separable (Kleinman-Bylander) form. Currently the following pseudopotential file formats are supported:
- Analytical Goedecker-Teter-Hutter (GTH) PSPs in the CP2K file format
- Numeric Unified Pseudopotential Format (UPF) PSPs
- Numeric pseudopotentials in the PSP8 (ABINIT) format.
In brief, the pseudopotential approach replaces the all-electron atomic potential with an effective atomic potential. In this pseudopotential, tightly-bound core electrons are completely eliminated ("frozen") and chemically-active valence electron wavefunctions are replaced with smooth pseudo-wavefunctions whose Fourier representations decay quickly. Both these transformations aim at reducing the number of Fourier modes required to accurately represent the wavefunction of the system, greatly increasing computational efficiency.
Different PSP generation codes produce various file formats which contain the same general quantities required for pesudopotential evaluation. GTH PSPs are constructed from a fixed functional form based on Gaussians, and the files simply tablulate various coefficients fitted for a given element. UPF and PSP8 PSPs take a more flexible approach where the functional form used to generate the PSP is arbitrary, and the resulting functions are tabulated on a radial grid in the file. The UPF file format is documented on the Quantum Espresso Website.
In this example, we will compare the convergence of an analytical GTH PSP with a modern numeric norm-conserving PSP in UPF format from PseudoDojo. Then, we will compare the bandstructure at the converged parameters calculated using the two PSPs.
using AtomsBuilder
using DFTK
using Unitful
using UnitfulAtomic
using PseudoPotentialData
using PlotsHere, we will use a Perdew-Wang LDA PSP from PseudoDojo, which is available via the JuliaMolSim PseudoPotentialData package. See the documentation of PseudoPotentialData for the list of available pseudopotential families.
family_upf = PseudoFamily("dojo.nc.sr.lda.v0_4_1.standard.upf");Such a PseudoFamily object acts like a dictionary from an element symbol to a pseudopotential file. They can be directly employed to select the appropriate pseudopotential when constructing an ElementPsp or a model based on an AtomsBase-compatible system. For the latter see the run_bands function below for an example.
An alternative to a PseudoFamily object is in all cases a plain Dict to map from atomic symbols to the employed pseudopotential file. For demonstration purposes we employ this in combination with the GTH-type pseudopotentials:
family_gth = PseudoFamily("cp2k.nc.sr.lda.v0_1.semicore.gth")
pseudopotentials_gth = Dict(:Si => family_gth[:Si])Dict{Symbol, String} with 1 entry:
:Si => "/home/runner/.julia/artifacts/966fd9cdcd7dbaba6dc2bf43ee50dd81e63e8837/Si.gth"First, we'll take a look at the energy cutoff convergence of these two pseudopotentials. For both pseudos, a reference energy is calculated with a cutoff of 140 Hartree, and SCF calculations are run at increasing cutoffs until 1 meV / atom convergence is reached.
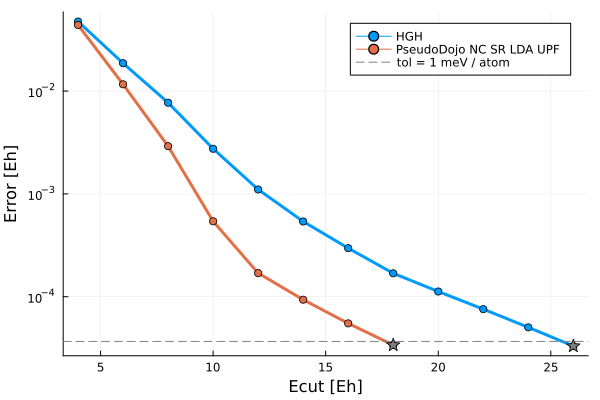
The converged cutoffs are 26 Ha and 18 Ha for the GTH and UPF pseudos respectively. We see that the GTH pseudopotential is much harder, i.e. it requires a higher energy cutoff, than the UPF PSP. In general, numeric pseudopotentials tend to be softer than analytical pseudos because of the flexibility of sampling arbitrary functions on a grid.
For some pseudopotentials the PseudoFamily contains hints for the recommended cutoffs as metadata. For most PseudoDojo potentials this is indeed the case:
recommended_cutoff(family_upf, :Si)(Ecut = 16.0, supersampling = 2.0, Ecut_density = 64.0)We see the recommended value is very close to what we determined above. Sometimes more detailed information is also available by looking at the raw metadata associated to this combination of pseudopotential family and element:
pseudometa(family_upf, :Si)Dict{String, Any} with 6 entries:
"rcut" => 10.0
"Ecut" => 16
"supersampling" => 2.0
"cutoffs_normal" => Dict{String, Any}("Ecut"=>16, "supersampling"=>2.0)
"cutoffs_high" => Dict{String, Any}("Ecut"=>22, "supersampling"=>2.0)
"cutoffs_low" => Dict{String, Any}("Ecut"=>12, "supersampling"=>2.0)Here, we see that multiple recommended cutoffs are made available in the metadata. Note, that recommended_cutoff can also be directly executed on a Model or an ElementPsp, e.g.
Si = ElementPsp(:Si, family_upf)
recommended_cutoff(Si)(Ecut = 16.0, supersampling = 2.0, Ecut_density = 64.0)pseudometa(Si)Dict{String, Any} with 6 entries:
"rcut" => 10.0
"Ecut" => 16
"supersampling" => 2.0
"cutoffs_normal" => Dict{String, Any}("Ecut"=>16, "supersampling"=>2.0)
"cutoffs_high" => Dict{String, Any}("Ecut"=>22, "supersampling"=>2.0)
"cutoffs_low" => Dict{String, Any}("Ecut"=>12, "supersampling"=>2.0)Next, to see that the different pseudopotentials give reasonably similar results, we'll look at the bandstructures calculated using the GTH and UPF PSPs. Even though the converged cutoffs are higher, we perform these calculations with a cutoff of 12 Ha for both PSPs.
function run_bands(pseudopotentials)
system = bulk(:Si; a=10.26u"bohr")
# These are (as you saw above) completely unconverged parameters
model = model_DFT(system; functionals=LDA(), temperature=1e-2, pseudopotentials)
basis = PlaneWaveBasis(model; Ecut=12, kgrid=(4, 4, 4))
scfres = self_consistent_field(basis; tol=1e-4)
bandplot = plot_bandstructure(compute_bands(scfres))
(; scfres, bandplot)
end;The SCF and bandstructure calculations can then be performed using the two PSPs, where we notice in particular the difference in total energies.
result_gth = run_bands(pseudopotentials_gth)
result_gth.scfres.energiesEnergy breakdown (in Ha):
Kinetic 3.1589991
AtomicLocal -2.1424274
AtomicNonlocal 1.6042947
Ewald -8.4004648
PspCorrection -0.2948928
Hartree 0.5515503
Xc -2.4000864
Entropy -0.0031626
total -7.926189861976result_upf = run_bands(family_upf)
result_upf.scfres.energiesEnergy breakdown (in Ha):
Kinetic 3.0953970
AtomicLocal -2.3650496
AtomicNonlocal 1.3082448
Ewald -8.4004648
PspCorrection 0.3952264
Hartree 0.5521686
Xc -3.1011585
Entropy -0.0032194
total -8.518855547593But while total energies are not physical and thus allowed to differ, the bands (as an example for a physical quantity) are very similar for both pseudos:
plot(result_gth.bandplot, result_upf.bandplot, titles=["GTH" "UPF"], size=(800, 400))