Modelling a gallium arsenide surface
This example shows how to use the atomistic simulation environment or ASE for short, to set up and run a particular calculation of a gallium arsenide surface. ASE is a Python package to simplify the process of setting up, running and analysing results from atomistic simulations across different simulation codes. For more details on the integration DFTK provides with ASE, see Atomistic simulation environment (ASE).
In this example we will consider modelling the (1, 1, 0) GaAs surface separated by vacuum.
Parameters of the calculation. Since this surface is far from easy to converge, we made the problem simpler by choosing a smaller Ecut and smaller values for n_GaAs and n_vacuum. More interesting settings are Ecut = 15 and n_GaAs = n_vacuum = 20.
miller = (1, 1, 0) # Surface Miller indices
n_GaAs = 2 # Number of GaAs layers
n_vacuum = 4 # Number of vacuum layers
Ecut = 5 # Hartree
kgrid = (4, 4, 1); # Monkhorst-Pack meshUse ASE to build the structure:
using ASEconvert
using PythonCall
a = 5.6537 # GaAs lattice parameter in Ångström (because ASE uses Å as length unit)
gaas = ase.build.bulk("GaAs", "zincblende"; a)
surface = ase.build.surface(gaas, miller, n_GaAs, 0, periodic=true);Get the amount of vacuum in Ångström we need to add
d_vacuum = maximum(maximum, surface.cell) / n_GaAs * n_vacuum
surface = ase.build.surface(gaas, miller, n_GaAs, d_vacuum, periodic=true);Write an image of the surface and embed it as a nice illustration:
ase.io.write("gaas_surface.png", surface * pytuple((3, 3, 1)), rotation="-90x, 30y, -75z")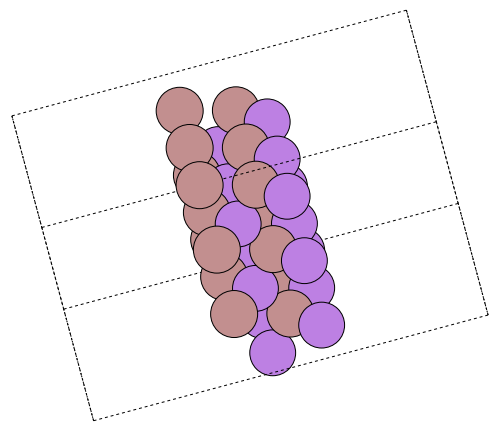
Use the pyconvert function from PythonCall to convert the ASE atoms to an AtomsBase-compatible system. This can then be used in the same way as other AtomsBase systems (see AtomsBase integration for details) to construct a DFTK model:
using DFTK
using PseudoPotentialData
pseudopotentials = PseudoFamily("cp2k.nc.sr.pbe.v0_1.largecore.gth")
model = model_DFT(pyconvert(AbstractSystem, surface);
functionals=PBE(),
temperature=1e-3,
smearing=DFTK.Smearing.Gaussian(),
pseudopotentials)Model(gga_x_pbe+gga_c_pbe, 3D):
lattice (in Bohr) : [7.55469 , 0 , 0 ]
[0 , 7.55469 , 0 ]
[0 , 0 , 40.0648 ]
unit cell volume : 2286.6 Bohr³
atoms : As₂Ga₂
pseudopot. family : PseudoFamily("cp2k.nc.sr.pbe.v0_1.largecore.gth")
num. electrons : 16
spin polarization : none
temperature : 0.001 Ha
smearing : DFTK.Smearing.Gaussian()
terms : Kinetic()
AtomicLocal()
AtomicNonlocal()
Ewald(nothing)
PspCorrection()
Hartree()
Xc(gga_x_pbe, gga_c_pbe)
Entropy()In the above we use the pseudopotential keyword argument to assign the respective pseudopotentials to the imported model.atoms. Try lowering the SCF convergence tolerance (tol) or try mixing=KerkerMixing() to see the full challenge of this system.
basis = PlaneWaveBasis(model; Ecut, kgrid)
scfres = self_consistent_field(basis; tol=1e-6, mixing=LdosMixing());n Energy log10(ΔE) log10(Δρ) Diag Δtime
--- --------------- --------- --------- ---- ------
1 -16.58723687264 -0.58 5.0 9.00s
2 -16.72865519175 -0.85 -1.01 1.0 5.55s
3 -16.73062490026 -2.71 -1.78 2.2 352ms
4 -16.73124084029 -3.21 -2.37 1.1 239ms
5 -16.73133686564 -4.02 -2.88 2.1 343ms
6 -16.73133977622 -5.54 -3.39 1.4 746ms
7 -16.73134016675 -6.41 -3.87 1.6 247ms
8 -16.73134019756 -7.51 -4.49 1.9 264ms
9 -16.73134020008 -8.60 -5.00 2.0 282ms
10 -16.73134020042 -9.47 -5.68 1.7 272ms
11 -16.73134020044 -10.78 -6.21 2.0 284ms
scfres.energiesEnergy breakdown (in Ha):
Kinetic 5.8593974
AtomicLocal -105.6100172
AtomicNonlocal 2.3494809
Ewald 35.5044300
PspCorrection 0.2016043
Hartree 49.5614317
Xc -4.5976637
Entropy -0.0000035
total -16.731340200435